
MptpB is an essential secreted virulence factor for M. tuberculosis. Inhibition of MptpB impairs mycobacterial survival in host macrophages and thus helps reduce tuberculosis infections. However, the binding mode of the biphenyl inhibitors, which are known as some of the most potent MptpB inhibitors, remains unclear. In this study, to understand the interactions between biphenyl inhibitors and MptpB, docking and molecular dynamics simulations were carried out using AutoDock and GROMACS softwares. Calculation results show that all the biphenyl inhibitors can be docked to the binding site of MptpB, with the acid warheads forming a hydrogen bond network at the active site. But the binding modes of other terminals of these inhibitors are different. The cyclohexyl and trifluoromethyl substituents at R1 and R2 sites are necessary for the inhibitors to adopt their double-site binding mechanism. The estimated binding affinities are basically consistent with the experimental results.MD simulations show that these binding complexes display different stability.
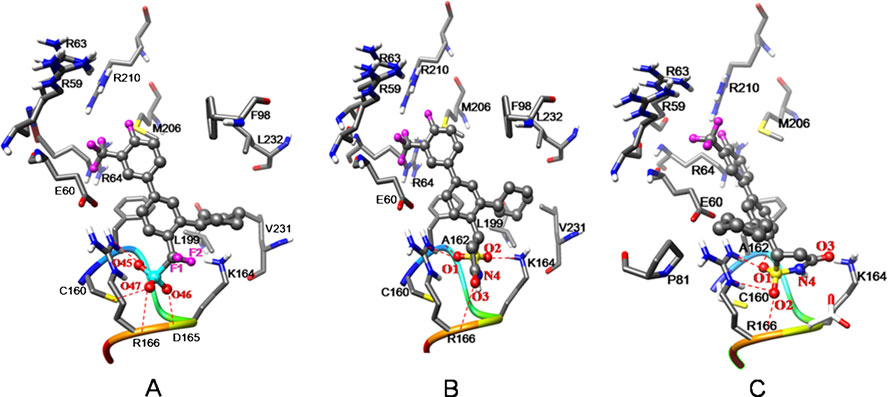
Docking conformations of MptpB complexes with difluoromethylphosphonic acid (DFMP) and isothiazolidinone (IZD) inhibitors. (a)
DFMP; (b) IZD_R and (C) IZD_S
Additional Information:
1. Author Information: Lihua Dong & Junyou Shi & Yongjun Liu
Correspondence: yongjunliu_1@sdu.edu.cn
2. Published : J Mol Model (2012) 18:3847–3856